EDMA form difference matrix
Peter Solymos
edma03-form-difference.RmdIntroduction
This tutorial explains how to calculate form difference based on 2 EDMA data objects with homologous landmarks. We will use 2 data sets: one with and the other without the mutation responsible for Crouzon syndrome:
library(EDMAinR)
#> EDMAinR 0.3-0 2023-08-21
file1 <- system.file("extdata/crouzon/Crouzon_P0_Global_MUT.xyz",
package="EDMAinR")
x1 <- read_xyz(file1)
file2 <- system.file("extdata/crouzon/Crouzon_P0_Global_NON-MUT.xyz",
package="EDMAinR")
x2 <- read_xyz(file2)Estimating the form matrices
We first estimate the mean forms (no bootstrap replicates are necessary).
Form matrices (\(FM\)) are formed as pairwise Euclidean distances between landmarks from EDMA fit objects using the estimated mean forms from objects \(A\) and \(B\).
Form difference
Form difference (\(FDM\)) is calculated as the ratio of form matrices (\(FM\)) from the numerator and denominator objects following Lele and Richtsmeier (1992, 1995): \(FDM(A,B) = FM(B)/FM(A)\).
Bootstrap distribution is based on fixing the reference \(FM\) and taking the ratio between the
reference \(FM\) and the bootstrap
\(FM\)s from the other object. The
ref_denom argument can be used to control which object is
the reference (it is the denominator by default), B is the
number of replicates drawn from this distribution:
fdm <- edma_fdm(numerator, denominator, B=25)
fdm
#> EDMA form difference matrix
#> Call: edma_fdm(numerator = numerator, denominator = denominator, B = 25)
#> 25 bootstrap runs (ref: denominator)
#> Tobs = 1.5981, p < 2.22e-16Global T-test
The global T-test is based on the pairwise distances in the \(FM\)s, taking the max/min ratio of the
distances. This is then done for all the B replicates,
which provides the null distribution.
Algorithm for global testing is as follows (suppose population 1 is the ‘reference’ population):
- Resample \(n_1\) observations from the first sample and compute \(FM_1^*\).
- Resample \(n_2\) observations from the first sample and compute \(FM_2^*\).
- Compute the \(FDM^* = FM_2^* / FM_1^*\) and \(T^* = max(FDM^*) / min(FDM^*)\).
- Repeat the above three steps
Btimes to get the p-value.
global_test provides a summary of the test,
plot_test helps to visualize the null distribution
(histogram) and the observed T statistic (vertical line):
global_test(fdm)
#>
#> Bootstrap based EDMA Tobs-test
#>
#> data: form difference matrix
#> Tobs -value = 1.5981, B = 25, p-value < 2.2e-16
plot_test(fdm)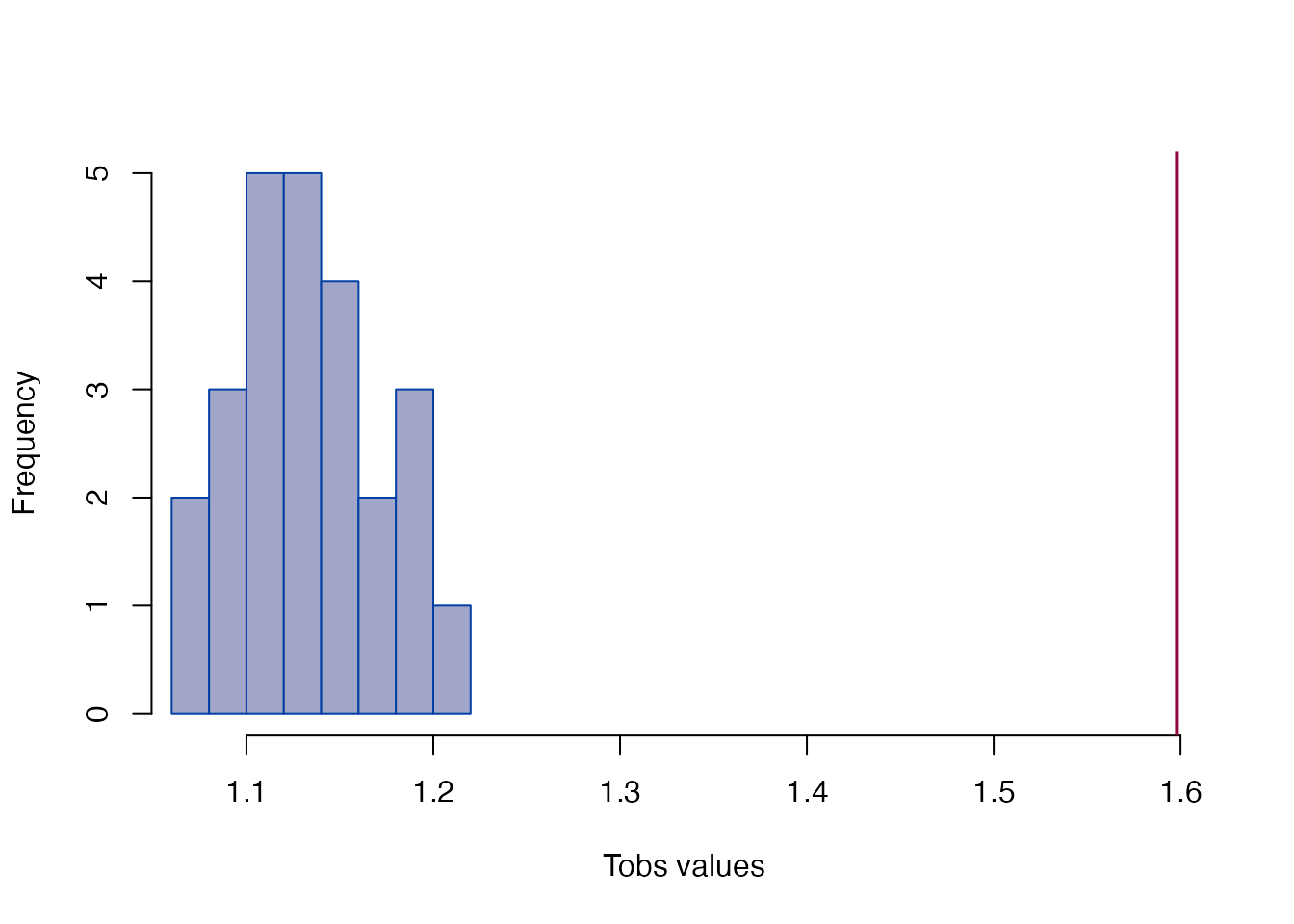
Local testing
The local testing is done based on the confidence intervals using the
stacked \(FDM\)s from the bootstrap.
Output structure is similar to the output of the get_fm
function, but the interpretation of the confidence intervals is
different due to the different nature of the bootstrap distribution.
Here the distribution characterizes the \(FDM\) and not the \(FM\). The confidence level can be changed
through the level argument.
The algorithm for confidence intervals for elements of the FDM is as follows:
- Resample \(n_1\) observations from the first sample and compute \(FM_1^*\).
- Resample \(n_2\) observations from the second sample and compute \(FM_2^*\).
- Compute the \(FDM* = FM_2^* / FM_1^*\).
- Repeat the above three steps B times to get the confidence intervals for the elements of the \(FDM\).
head(get_fdm(fdm))
#> row col dist lower upper
#> 1 bas amsph 1.0425379 1.0291234 1.0572437
#> 2 cpsh amsph 0.9823765 0.9616780 0.9988809
#> 3 ethma amsph 1.0037761 0.9914403 1.0156289
#> 4 ethmp amsph 0.9401242 0.9278436 0.9510568
#> 5 laalf amsph 0.9878983 0.9788081 0.9990751
#> 6 lasph amsph 1.0161844 0.9891307 1.0317053
head(get_fdm(fdm, sort=TRUE, decreasing=TRUE))
#> row col dist lower upper
#> 136 ethmp ethma 1.377873 1.279108 1.479039
#> 697 rpto lpto 1.131002 1.087753 1.164933
#> 179 laalf ethmp 1.095672 1.070444 1.124045
#> 200 raalf ethmp 1.094267 1.067043 1.125650
#> 881 rpmx raalf 1.080811 1.061278 1.103610
#> 607 rpns lpns 1.078698 1.058532 1.122745
head(get_fdm(fdm, sort=TRUE, decreasing=FALSE))
#> row col dist lower upper
#> 527 lsqu lpfl 0.8622059 0.8089977 0.9459507
#> 1022 rsqu rpfl 0.8781312 0.8243285 0.9393457
#> 93 ethmp cpsh 0.9034404 0.8810750 0.9250952
#> 212 rpsh ethmp 0.9177018 0.8965352 0.9395801
#> 190 lpsh ethmp 0.9181652 0.8947758 0.9348729
#> 4 ethmp amsph 0.9401242 0.9278436 0.9510568Differences below or above the confidence limits indicate distances
that significantly deviate from the bootstrap based null expectation,
and thus are related to landmarks that drive the differences. Inspecting
the highest and lowest differences (using sort) can help
revealing these landmarks.
The lower and upper limits of the
confidence intervals are based on confint (row names
indicate the unsorted sequence of landmark pairs as in the output from
get_fdm(fdm)):
head(confint(fdm))
#> 2.5% 97.5%
#> bas-amsph 1.0291234 1.0572437
#> cpsh-amsph 0.9616780 0.9988809
#> ethma-amsph 0.9914403 1.0156289
#> ethmp-amsph 0.9278436 0.9510568
#> laalf-amsph 0.9788081 0.9990751
#> lasph-amsph 0.9891307 1.0317053The plot_ci function shows the pairwise differences and
confidence intervals. The x-axis label shows the landmark pairs, and
gets really busy. Use the xshow=FALSE argument to remove
labels.
plot_ci(fdm, xshow=FALSE)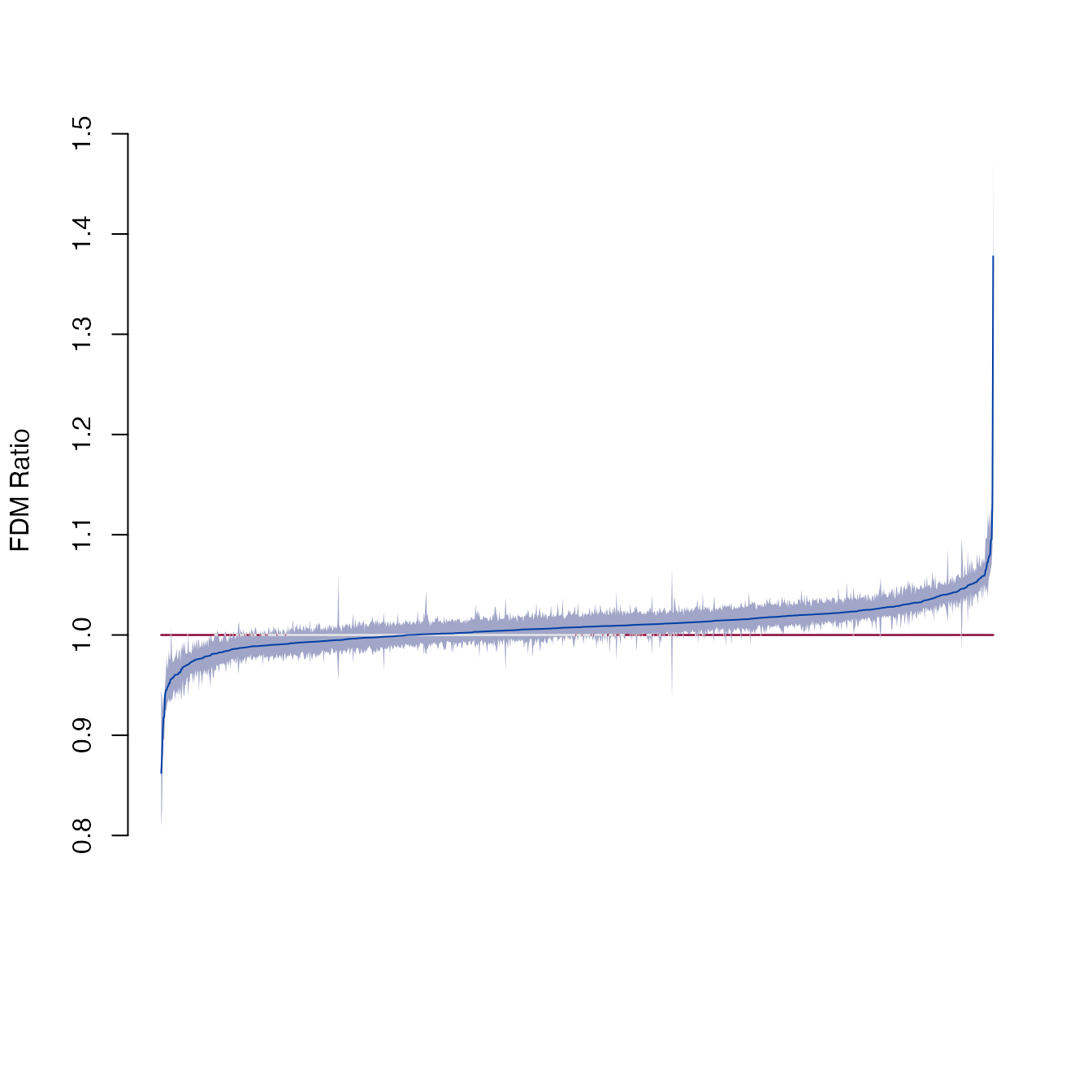
We can use the stacked \(FDM\) data frame to make a ggplot2 based plot:
library(ggplot2)
df <- get_fdm(fdm, sort=TRUE)
df$x <- 1:nrow(df) # make x-axis continuous
df$name <- paste(df$row, df$col) # add names
p <- ggplot(data=df, aes(x=x, y=dist,
ymin=lower, ymax=upper, label=name)) +
geom_ribbon(col="#0000ff40", fill="#0000ff40") +
geom_line() +
geom_hline(yintercept=1, col=2) +
labs(y="FDM Ratio", x="Landmark Pairs") +
theme_linedraw() +
theme(axis.text.x=element_blank())
p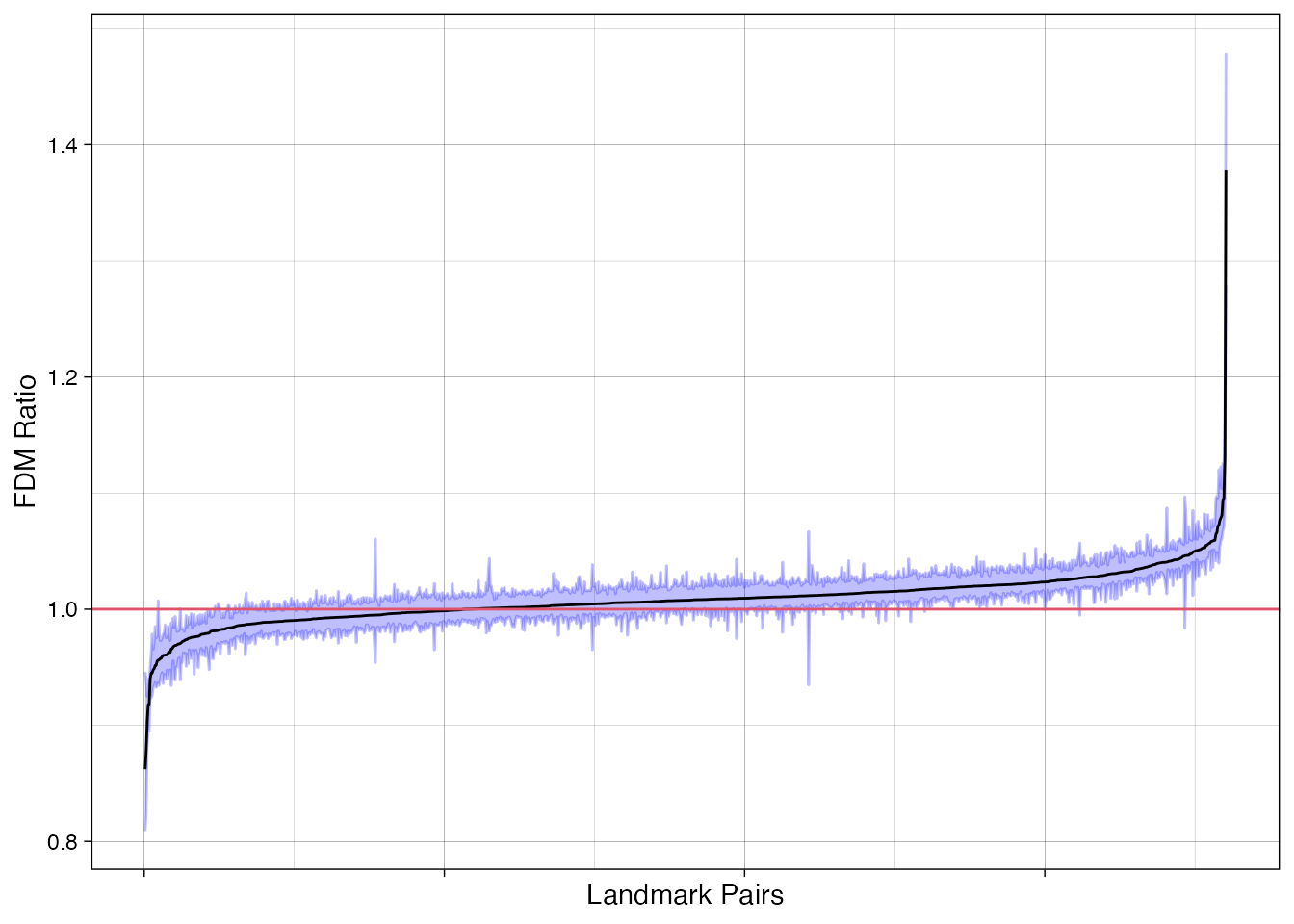
Once we have a ggplot2 version of a plot, we can load the plotly package to make the object interactive:
Influential landmarks
We can consider a landmark influential with respect to the form difference if after removing the landmark, the global T value moves close to 1. Such a case would indicate that the landmark in question is driving the form differences (i.e. pairwise distances between the landmark and other landmarks contribute to the maximum value in the T statistic). If, however, we remove a non-influential landmark, we expect the T value not to change a lot. Therefore, the ‘drop’ in the T value after removing a landmark can be used to rank landmarks based on their influence.
Influential landmarks are identified by leaving one landmark out at a time, then calculating the the T-statistic based on the remaining distances. We can use the bootstrap distribution to see of the T value ‘drop’ makes the global test non-significant. This means that after removing the landmark, the form difference cannot be distinguished from the null expectation.
The most influential landmark is the one with the largest drop in T
value compared to the original T statistic. Tdrop is the
newly calculated T value after leaving out the landmark in question:
infl <- get_influence(fdm)
head(infl[order(infl$Tdrop),], 10)
#> landmark Tdrop lower upper
#> 4 ethma 1.311754 1.074204 1.253519
#> 5 ethmp 1.311754 1.074204 1.253519
#> 14 lpfl 1.569097 1.074204 1.350022
#> 21 lsqu 1.569097 1.074204 1.350022
#> 1 amsph 1.598078 1.074204 1.360890
#> 2 bas 1.598078 1.074204 1.360890
#> 3 cpsh 1.598078 1.074204 1.360890
#> 6 laalf 1.598078 1.074204 1.360890
#> 7 lasph 1.598078 1.074204 1.360890
#> 8 lflac 1.598078 1.074204 1.360890Note: we used the
orderfunction to create an index to order the rows of theinfldata frame.get_influencealso takes alevelargument for specifying the desired confidence interval (default is 95%).
The plot function shows the landmarks ordered by
Tdrop with the influential landmarks on the left-hand side
of the plot. The horizontal line on the top indicates the original T
value (with all the landmarks considered), the increasing line shows
Tdrop, the shaded area is the null distribution for the T
statistic:
plot(infl)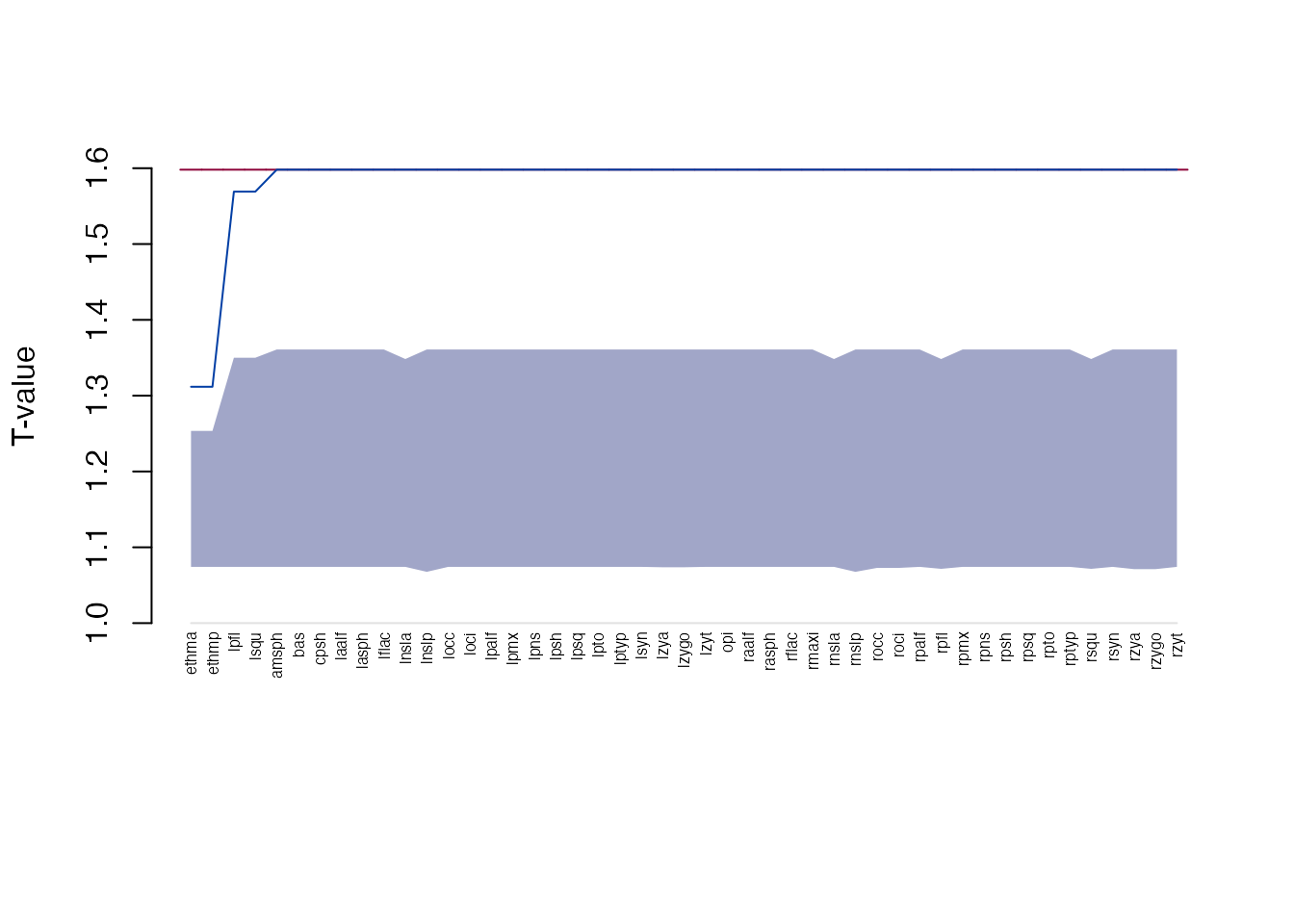
Here is the ggplot2 version:
df <- infl[order(infl$Tdrop),]
df$landmark <- factor(as.character(df$landmark), as.character(df$landmark))
p <- ggplot(data=df, aes(x=landmark, y=Tdrop, ymin=lower, ymax=upper, group=1)) +
geom_ribbon(col="#0000ff40", fill="#0000ff40") +
geom_hline(yintercept=global_test(fdm)$statistic, col=2) +
geom_line() +
labs(y="T-value", x="Landmarks") +
theme(axis.text.x=element_text(angle = 45,
vjust = 1, hjust=1))
p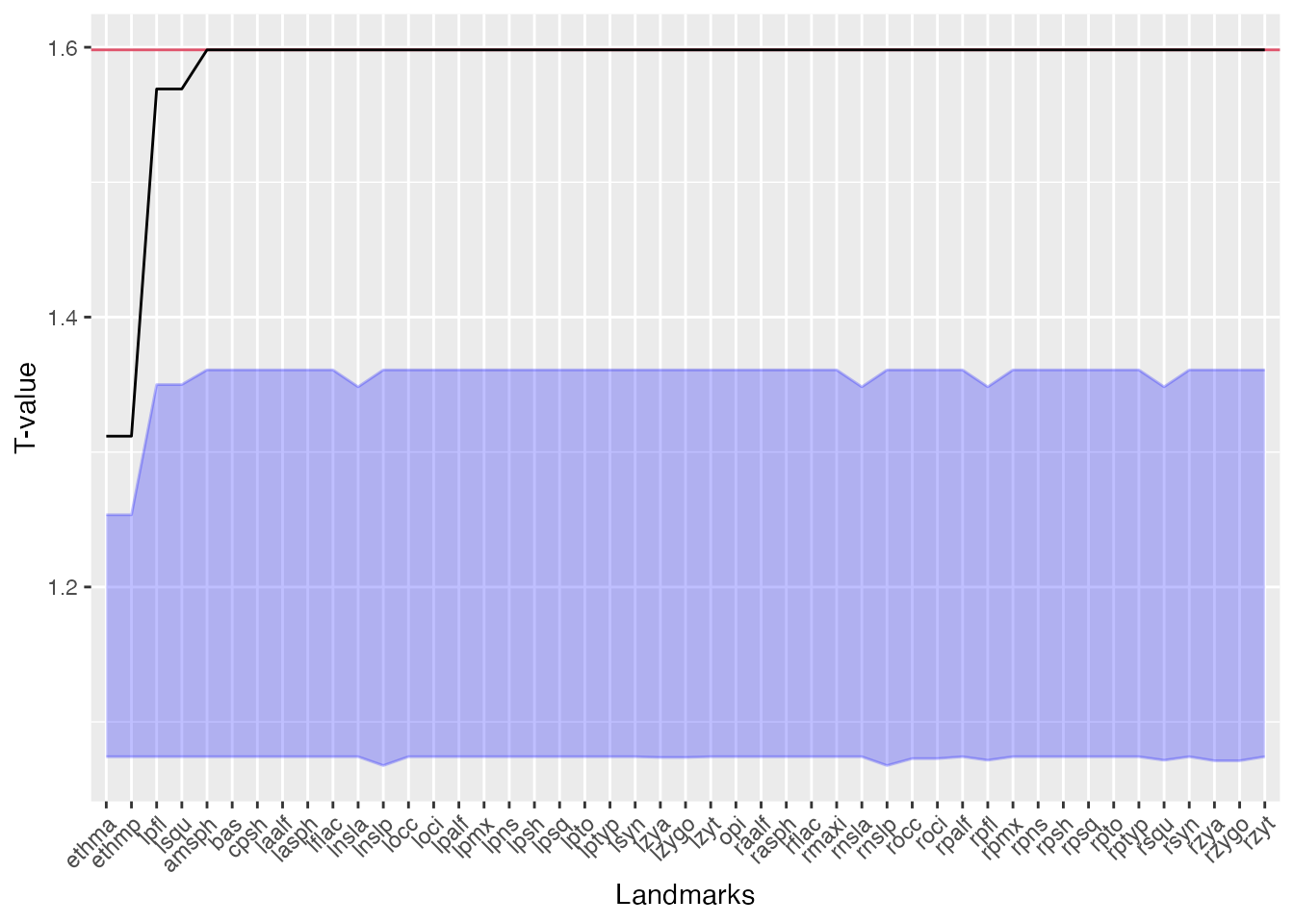
An the interactive version:
ggplotly(p)Ordination and clustering for specimens
The ordination and cluster dendrogram shows the two sets of specimens
from the 2 objects in the same diagram. The 2 sets are combined with the
combine_data function:
x1
#> EDMA data: Crouzon P0 MUT
#> 47 landmarks, 3 dimensions, 28 specimens
x2
#> EDMA data: Crouzon P0 UNAFF
#> 47 landmarks, 3 dimensions, 31 specimens
(x12 <- combine_data(x1, x2))
#> EDMA data: data with 2 groups
#> 47 landmarks, 3 dimensions, 59 specimens
table(x12$groups)
#>
#> 1 2
#> 28 31The visualization otherwise use the same principles as described for
EDMA data objects. The difference is that the specimens and their labels
are colored (using a the default qualitative palette) according to the
groups (numerator vs. denominator).
If the the numerator and denominator objects are different (global T value is high, \(p\) value is low, there are influential landmarks), we expect the two groups to separate in ordination space and along the dendrogram:
plot_ord(fdm)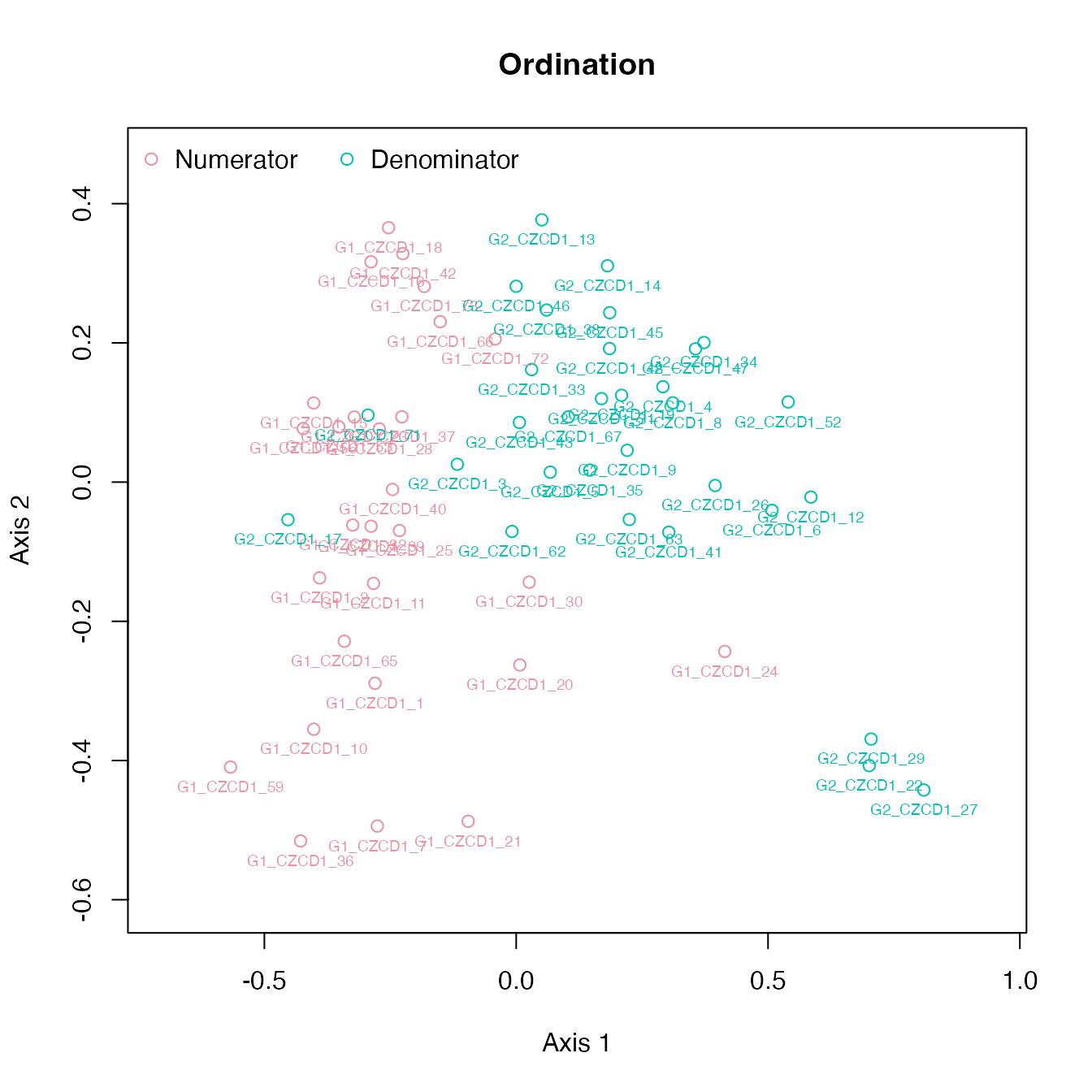
The dendrogram leaves (specimen labels) are also colored by groups:
plot_clust(fdm)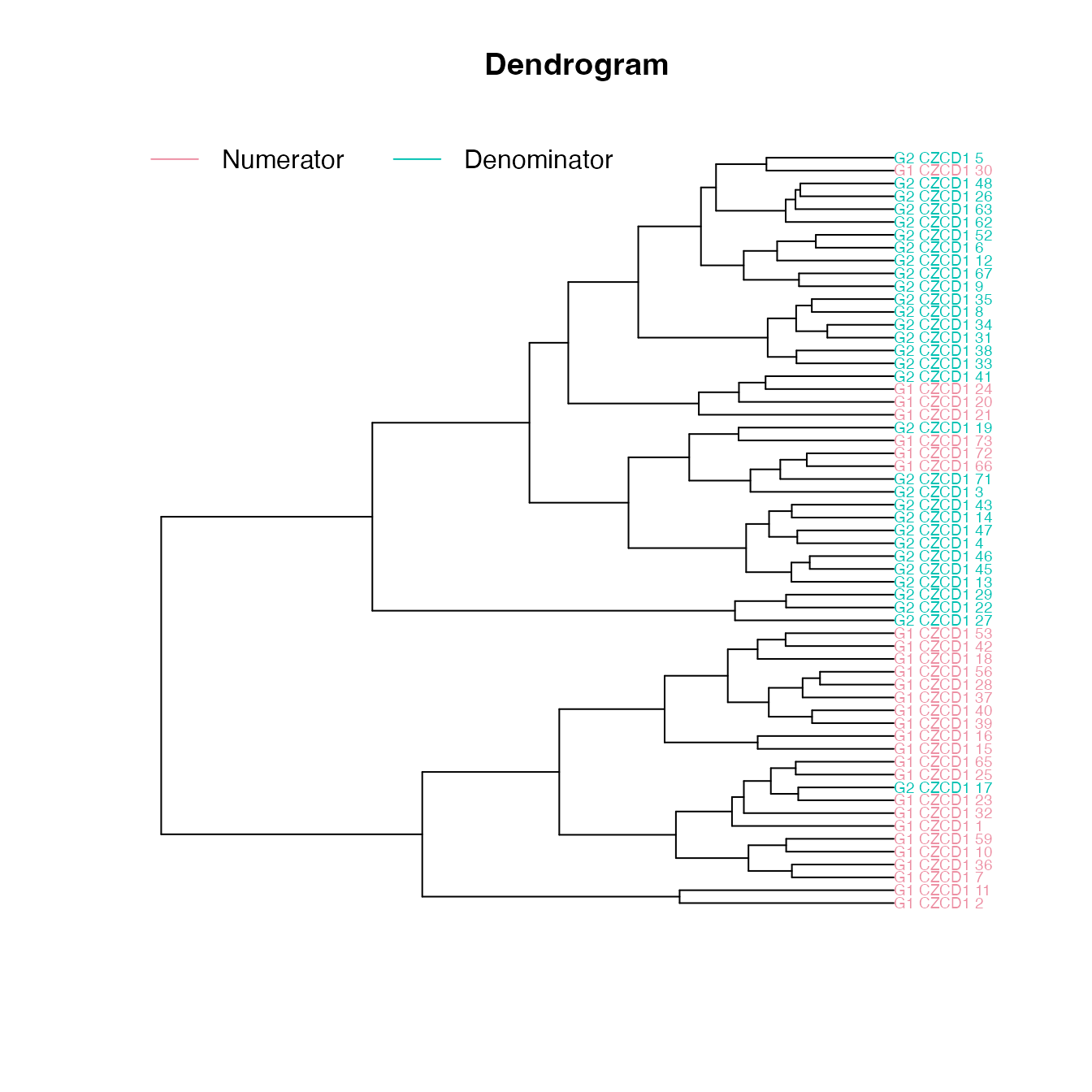
The colors can be changed via the color options:
options(op)Visualizing landmarks
The 2D and 3D plots produce a plot of the mean form from the
reference object (‘prototype’). The color intensity for the landmarks
(dots) is associated with the Tdrop influence value (larger
the difference, the more intensive the color; red by default). Lines
between the landmarks represent distances. We use the diverging
palettes: <1 differences are colored blue (1st half of the palette),
>1 differences are colored red (2st half of the palette).
plot_2d(fdm, cex=2)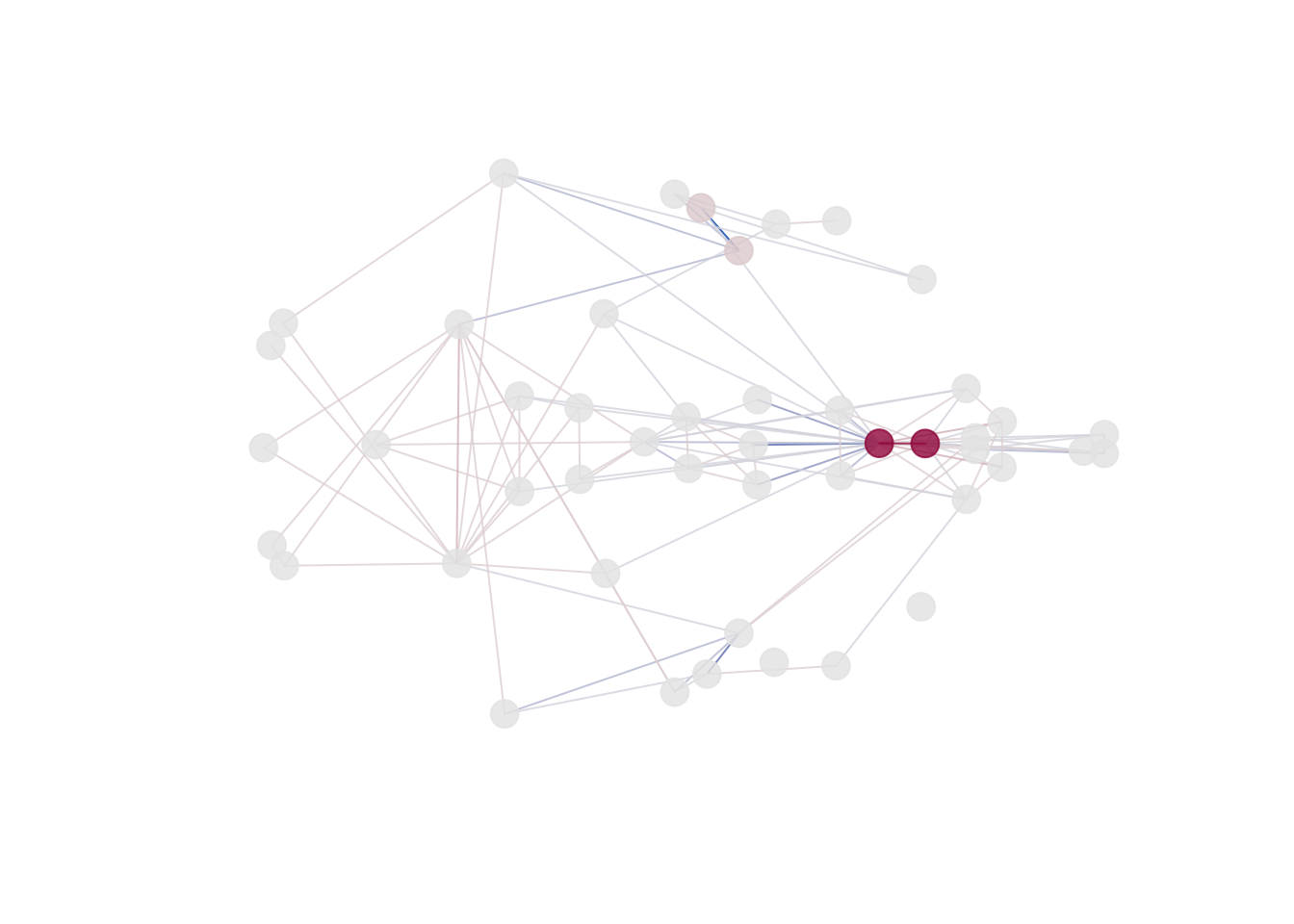
library(rgl)
xyz <- plot_3d(fdm)
text3d(xyz, texts=rownames(xyz), pos=1) # this adds names
decorate3d() # this adds the axes
rglwidget(width = 600, height = 600, reuse = FALSE)The 2D and 3D plots do not display all the pairwise distances. If
displaying all edges is desired, use the all=TRUE
argument.